https://academic.oup.com/bib/article/24/1/bbac581/6986359
RNAdegformer: accurate prediction of mRNA degradation at nucleotide resolution with deep learning
Abstract. Messenger RNA-based therapeutics have shown tremendous potential, as demonstrated by the rapid development of messenger RNA based vaccines for CO
academic.oup.com
요약
메신저 RNA(mRNA) 기반 치료제는 COVID-19에 대한 mRNA 백신의 빠른 개발을 통해 엄청난 잠재력을 보여주었습니다. 그러나 mRNA의 고유한 열적 불안정성, 특히 화학적 분해 반응인 인라인 가수분해로 인해 전 세계적으로 mRNA 백신의 배포가 어려워졌습니다. 따라서 RNA 분해를 예측하고 이해하는 것이 중요한 과제가 되었습니다. 이 연구에서는 RNAdegformer라는 효과적이고 해석 가능한 모델 구조를 제시하며, 이는 RNA 분해를 예측하는 데 뛰어난 성능을 발휘합니다. RNAdegformer는 RNA 시퀀스를 처리할 때, 컴퓨터 비전 및 자연어 처리 분야에서 지배적인 기술로 입증된 자기 주의(Self-Attention)와 컨볼루션(Convolution)이라는 두 가지 딥러닝 기법을 활용하며, RNA의 생물물리학적 특징을 고려합니다. 우리는 RNAdegformer가 COVID-19 mRNA 백신에 대한 뉴클레오타이드 해상도의 분해 특성을 예측하는 데 이전의 최고 방법들보다 우수한 성능을 보인다는 것을 입증했습니다. RNAdegformer의 예측은 이전의 최고 방법들과 비교했을 때 RNA의 시험관 내 반감기와의 상관관계도 개선되었습니다. 또한, 우리는 자기 주의 맵의 직접적인 시각화를 통해 정보에 기반한 의사결정을 도울 수 있음을 보여주었습니다. 나아가, 우리의 모델은 leave-one-feature-out 분석을 통해 mRNA 분해 속도를 결정하는 중요한 특징들을 밝혀냈습니다.
키워드: mRNA 백신 분해, 딥러닝, 생물정보학, COVID-19 mRNA
서론
메신저 RNA(mRNA) 치료제는 모듈성을 제공하고 이론적으로는 어떤 단백질이든 전달하고 번역할 수 있는 매우 유망한 플랫폼으로 떠오르고 있습니다. 포유동물 세포주에서 발현된 재조합 단백질과 비교할 때, mRNA는 시험관 내 전사를 통해 더 빠르고 유연하게 생산될 수 있으며, COVID-19에 대한 mRNA 기반 백신의 신속한 배포는 mRNA 치료제의 잠재력을 입증합니다. 그럼에도 불구하고 mRNA 기반 치료제는 mRNA 분자의 고유한 불안정성이라는 근본적인 한계에 직면해 있습니다. 그 결과, mRNA 백신은 시험관 내 및 생체 내에서 RNA 불안정성으로 인해 여전히 효능이 감소하는 문제를 겪고 있습니다. RNA의 분해는 분자가 인라인 가수분해에 얼마나 취약한지에 따라 달라지지만, 현재로서는 특정 mRNA의 골격 중 어느 부분이 가수분해에 취약하고 어느 부분이 분해로부터 안전한지에 대해 많이 알려져 있지 않습니다. mRNA 분해를 이해하면 더 열 안정성이 높은 RNA 치료제를 설계할 수 있어 배포의 형평성을 높이고 비용을 절감하며, 심지어 효과를 증가시킬 수 있습니다. 또한, 확률적 최적화를 통해 동일한 단백질을 같은 양으로 번역하는 더 열 안정성이 높은 mRNA 서열을 설계할 수 있다는 것도 입증되었습니다.
mRNA의 이차 구조는 mRNA의 안정성과 번역과 긍정적인 상관관계가 있는 것으로 나타났으며, 이차 구조를 최적화하여 반감기와 번역 효율을 증가시키는 것이 가능한 것으로 밝혀졌습니다. 많은 동적 프로그래밍 기반의 RNA 이차 구조 패키지는 mRNA 서열의 이차 구조를 합리적인 정확도로 예측할 수 있습니다. 그러나 RNA 이차 구조 패키지는 상당히 이상적이며, mRNA의 분해는 단순한 이차 구조뿐만 아니라 mRNA 서열의 지역적 및 전역적 맥락에도 영향을 받을 수 있습니다. 따라서 mRNA 분해를 연구하기 위해 대체 경로를 탐색하는 것이 중요합니다.
딥러닝은 데이터 기반의 모델링 접근 방식으로, 이미지 인식, 자연어 처리, 유전체학 및 계산 생물학을 포함한 많은 분야에서 성공을 거두고 있습니다. 이 기술은 대규모 데이터셋에 대한 신경망 학습을 통해 DNA/RNA 서열의 기능, 기원 및 특성을 효율적으로 예측하는 데 사용되고 있습니다. 하지만 이러한 순차적 계산 접근 방식은 병렬화가 어렵고, 장거리 의존성을 다루는 데 한계가 있습니다. 컨볼루션 신경망(CNN)은 모티프 인식에 능숙하며, 기존의 비딥러닝 접근 방식보다 더 나은 성능을 제공하는 것으로 나타났습니다. 그러나 DNA/RNA 작업에 필수적인 장거리 의존성을 캡처하는 데 여전히 어려움을 겪고 있습니다. 그래프 기반 모델도 RNA 결합 연구에 적용되었습니다. 반면, 트랜스포머는 입력 또는 출력 시퀀스에서 거리에 상관없이 의존성을 모델링할 수 있는 주의 메커니즘에만 의존하는 새로운 구조로 제안되었습니다. 트랜스포머는 자연어 처리 작업에서 큰 성공을 거두었으며, 최근에는 DNA 서열 분류에도 사용되었습니다. 그러나 RNA 서열의 생물물리학적 특성을 연구하는 데 트랜스포머를 사용하는 연구는 드뭅니다.
본 연구에서는 컨볼루션과 자기 주의를 결합하여 지역적 및 전역적 의존성을 모두 포착할 수 있는 신경망 모델 RNAdegformer를 제시합니다. 이 모델은 mRNA 서열의 분해 특성을 예측하는 데 있어 높은 정확도와 해석 가능성을 제공합니다. 우리는 21일간의 OpenVaccine 챌린지에 참여하여 RNAdegformer를 훈련시키기 위해 데이터를 사용했습니다. 이중 가닥 DNA가 상보적 염기 간의 수소 결합을 형성하는 반면, 단일 가닥 RNA는 스스로 이차 구조를 형성하여 RNA 분자를 안정화하는 것으로 알려져 있습니다. 따라서 우리는 RNA 이차 구조에 대한 기존 생물물리학적 모델을 사용하여 딥러닝 모델에 지식을 주입하고 RNA 분해를 예측합니다. 감독 학습, 비감독 학습 및 반감독 학습과 같은 고급 학습 기법을 결합하여 RNAdegformer가 주어진 서열의 각 위치에서 RNA 분해율을 예측하는 데 이전 최고 방법들을 능가함을 입증합니다. 이는 안정적인 mRNA 백신 및 치료제를 예측하고 생산하는 데 중요한 작업입니다. 또한, RNAdegformer는 다른 기계 학습 및 동적 프로그래밍 알고리즘과 비교할 때, 훈련 데이터셋보다 훨씬 긴 서열의 반감기를 예측하는 데 있어 더 나은 일반화를 보여줍니다. 마지막으로, RNAdegformer는 leave-one-feature-out(LOFO) 테스트를 통해 mRNA 분해 예측에서 중요한 특징을 밝혀내어 RNA 분해에 대한 이해를 증진시킵니다.
방법론
OpenVaccine 챌린지 데이터셋
Eterna 커뮤니티에서 주최한 OpenVaccine 챌린지[46]는 Kaggle 참가자들의 데이터 과학 전문 지식을 결집하여 mRNA 분해를 정확하게 예측할 수 있는 모델을 개발하는 것을 목표로 했습니다. 21일간의 챌린지 동안 참가자들은 2400개의 107 염기쌍(bp) mRNA 서열을 제공받았으며, 이 중 첫 68 염기쌍은 각 위치에서 다섯 가지 분해 특성에 대한 레이블이 지정되었습니다. 이러한 특성은 반응성(reactivity), pH 10에서의 분해(deg_pH10), pH 10에서 Mg와 함께하는 분해(deg_Mg_pH10), 50°C에서의 분해(deg_50C) 및 50°C에서 Mg와 함께하는 분해(deg_Mg_50C)입니다. 이들 특성에 대한 자세한 내용은 이 링크에서 확인할 수 있습니다.
대부분의 Kaggle 대회와 마찬가지로, 테스트 세트는 공공 테스트 세트와 비공개 테스트 세트로 나뉘었습니다. 대회 기간 동안 공공 테스트 세트의 결과는 확인할 수 있었지만, 비공개 테스트 세트의 결과는 감춰져 있었습니다(Figure 1A). 훈련 데이터와 공공 테스트 세트를 얻기 위한 실험 절차는 [7]에 자세히 설명되어 있습니다. 최종 평가는 130 염기쌍으로 이루어진 3005개의 mRNA 서열로 구성된 비공개 테스트 세트의 일부에서 수행되었으며, 이들의 분해 측정은 21일 챌린지 기간 동안 수행되었고 대회 종료 시 공개되었습니다. 테스트 세트는 다음 세 가지 기준에 따라 선별되었습니다.
- 다섯 가지 분해 특성 중 최소값이 -0.5보다 커야 합니다.
- 다섯 가지 분해 특성에 걸친 평균 신호/노이즈 비율이 1.0보다 커야 합니다. [신호/노이즈는 평균(68개의 뉴클레오타이드에서 측정된 값)/평균(68개의 뉴클레오타이드에서 측정된 값의 통계적 오차)로 정의됩니다].
- 서열은 유사성이 50% 미만인 군집으로 나누어졌으며, 군집당 3개 이하의 구성원을 가진 군집에서 선택되었습니다.

Figure 1 연구에서 사용된 데이터셋
A. OpenVaccine 데이터셋 시각화
B. eGFP, 나노루시퍼라제(Nanoluciferase), 그리고 MEV 서열로 구성된 시험관 내 반감기 데이터셋 시각화
선별 후, 테스트 세트에는 1172개의 서열만 남았습니다. 최종 평가는 다섯 가지 특성 중 세 가지(반응성, deg_Mg_pH10 및 deg_Mg_50C)에 대해 수행되었습니다. 훈련 세트와 달리, 테스트 세트에는 더 긴 mRNA 서열, 더 다양한 서열, 그리고 서열당 더 많은 측정치(처음 91개의 위치)가 포함되었습니다. 실제로, 테스트 세트에서는 훈련 샘플보다 더 많은 예측이 필요했습니다. 대회에서 순위를 매기는 데 사용된 지표는 열별 평균 제곱근 오차(MCRMSE, Mean Columnwise Root Mean Squared Error)입니다.

여기서 M은 열의 수, N은 예측된 위치의 수, y_i는 실제 값(ground truth), 그리고 는 예측된 값(predicted value)입니다. 추가적으로, 추가적인 분석에서 R^2 점수(결정 계수, coefficient of determination)를 사용합니다.
시험관 내 반감기 데이터셋
OpenVaccine 데이터셋 외에도, 우리는 세 가지 CDS 그룹(eGFP, 나노루시퍼라제(Nanoluciferase), 그리고 짧은 다중 에피토프 백신(MEV))의 시험관 내 반감기에 대한 최근 이용 가능한 데이터셋을 독립적인 테스트를 위해 사용합니다[7]. 이 데이터셋은 69개의 Nanoluc CDS 변이체, 13개의 eGFP 변이체, 그리고 9개의 MEV 변이체로 구성되어 있습니다(Figure 1B). 이들 서열은 UTR 영역을 포함한 전체 길이의 mRNA 서열임을 주목해야 합니다. 우리의 모델이 직접적으로 반감기를 출력하지 않기 때문에, 우리는 반감기의 대용치로서 CDS 영역의 반응성 예측값을 합산하고, 반응성 합계와 반감기의 피어슨 상관계수(Pearson R correlation)를 계산하여 모델의 성능을 평가합니다.
-----------------------------------------------------------------------------------------------------------------------------------------------------------------
반응성 합계와 반감기의 피어슨 상관계수(Pearson R correlation)는 두 변수 간의 상관 관계를 측정하는 통계적 방법입니다. 이 경우, 반응성 합계는 특정 mRNA 서열의 CDS(코딩 서열) 영역에서 예측된 반응성 값을 모두 더한 것입니다. 반감기는 mRNA가 분해되어 절반으로 감소하는 데 걸리는 시간, 즉 mRNA의 안정성을 나타내는 지표입니다.
피어슨 상관계수는 -1에서 1 사이의 값을 가지며, 이 값이 1에 가까울수록 두 변수 간의 양의 선형 상관 관계가 강하다는 것을 의미합니다. 반대로, -1에 가까울수록 음의 선형 상관 관계가 강하다는 것을 의미하며, 0에 가까울수록 상관 관계가 거의 없다는 것을 의미합니다.
이 연구에서 피어슨 상관계수를 사용한 이유는 모델이 예측한 반응성 합계와 실제 측정된 mRNA 반감기 사이의 관계가 얼마나 강한지를 평가하기 위해서입니다. 상관계수가 높을수록 모델이 mRNA의 반감기를 잘 예측하고 있다는 것을 의미합니다.
-----------------------------------------------------------------------------------------------------------------------------------------------------------------
K-머와 1차원 컨볼루션(K-mers with 1-D Convolutions)
RNAdegformer는 각 뉴클레오타이드를 임베딩한 후, 1차원 컨볼루션을 사용하여 K-머를 추출함으로써 지역적 의존성을 포착합니다. RNA 서열에서 K-머를 추출하는 과정은 서열의 한쪽 끝에서 다른 쪽 끝으로 한 번에 한 위치씩 이동하면서 서열의 스냅샷을 찍는 크기 k의 슬라이딩 윈도우로 생각할 수 있으며, 이는 딥러닝에서 사용되는 컨볼루션 연산과 개념적으로 동일합니다. 다음은 벡터 v에 대해 컨볼루션을 적용하는 간단한 예시입니다. 여기서 n은 벡터의 길이이고, 컨볼루션 커널 w는 벡터 v에 대해 컨볼루션을 수행합니다. 만약 컨볼루션 커널이 한 번에 한 위치씩 이동한다면, 다음과 같은 점곱의 출력 벡터 z가 계산됩니다:

K_i는 컨볼루션 가중치 행렬의 번째 요소이고, S_{p+i}는 입력 벡터의 p+i번째 요소이며, p는 출력 벡터 O에서 위치를 나타냅니다. 이 경우, 컨볼루션 연산은 한 번에 세 위치의 지역 정보를 집계하므로, 만약 가 뉴클레오타이드 서열이라면, 컨볼루션 연산은 본질적으로 DNA 서열에서 3-머(3-mer)를 추출하는 것입니다.
우리 모델은 RNA 뉴클레오타이드 서열을 입력으로 받기 때문에, 먼저 각 뉴클레오타이드를 고정 크기 d_{model}의 임베딩으로 변환합니다. 따라서 각 서열에 대해 I∈{R}^{l × d_{model}}의 텐서가 생성되며, 여기서 l은 서열의 길이를 나타냅니다. 이제 K-머를 만들기 위해, 우리는 패딩 없이 스트라이드 = 1로 텐서 I에서 컨볼루션을 수행합니다. 커널 크기 k를 가진 컨볼루션 연산이 I에 대해 수행되면, K-머 서열을 나타내는 새로운 텐서 k∈R^{(l-k+1) × d_{model}}가 생성됩니다. 마지막으로, 각 K-머는 크기 d_{model}의 특성 벡터로 표현됩니다. 1D 컨볼루션 레이어 다음에는 항상 레이어 정규화 레이어가 뒤따릅니다 [47].
사실, 우리의 K-머 표현은 딥러닝에서 전통적인 단어 표현과 다릅니다. 전통적인 방식에서는 어휘 내의 각 단어가 룩업 테이블에서 특성 벡터에 직접 대응합니다. K-머에 대해 룩업 테이블을 사용하는 단점은 OpenVaccine 데이터셋에 존재하는 모든 가능한 K-머 중 매우 적은 비율만이 포함되어 있으며, 네트워크가 보지 못한 K-머에 대해 일반화하기가 거의 불가능하다는 점입니다. 게다가, 더 큰 크기의 K-머를 임베딩으로 표현하려면 엄청난 양의 매개변수가 필요합니다. 주어진 k에 대해 가능한 총 K-머 수가 4^k이기 때문입니다. 예를 들어, 우리가 컨볼루션으로 생성한 가장 큰 K-머는 9개의 뉴클레오타이드로 이루어져 있으며, 이를 표현하기 위해 임베딩을 사용하려면 262,144개의 크기(우리의 경우 256)의 임베딩이 필요합니다.
-----------------------------------------------------------------------------------------------------------------------------------------------------------------
- 주어진 k에 대해 가능한 총 K-머 수가 4^k인 이유:
RNA에는 4가지 뉴클레오타이드(A, U, G, C)가 있습니다. k-머는 k개의 연속된 뉴클레오타이드를 의미합니다.
- 각 위치에서 4가지 선택지가 있습니다.
- 이 선택을 k번 반복합니다.
따라서 가능한 모든 조합의 수는 4의 k제곱, 즉 4^k가 됩니다.
예를 들어:
- 1-머 (k=1): 4^1 = 4 가지 (A, U, G, C)
- 2-머 (k=2): 4^2 = 16 가지 (AA, AU, AG, AC, UA, UU, ...)
- 3-머 (k=3): 4^3 = 64 가지
- 9-머 (k=9): 4^9 = 262,144 가지
이 때문에 k가 커질수록 가능한 k-머의 수가 급격히 증가하여, 전통적인 임베딩 방식으로는 메모리 사용량이 매우 커지게 됩니다.
-----------------------------------------------------------------------------------------------------------------------------------------------------------------
Transformer 인코더





RNA 분해 예측을 위한 생물물리 모델 통합
RNA 분해를 정확하게 예측하려면 단순히 서열 정보만으로는 부족하며, 여기에서는 RNAdegformer를 훈련할 때 생물물리 모델을 추가적인 특징으로 활용하는 방법을 설명합니다. Viennafold와 같은 생물물리 모델은 동적 프로그래밍과 열역학적 채점 함수를 사용하여 RNA 이차 구조를 예측합니다. 이 모델들은 직접적으로 RNA 분해를 예측할 수는 없지만, 이차 구조 예측은 신경망을 훈련하여 분해를 직접 예측하는 데 매우 유용합니다.

여기서 γ는 학습 가능한 파라미터이고, M_{bp}는 수정된 염기쌍 결합 확률 행렬(알고리즘 1)입니다. 원래의 염기쌍 결합 확률 행렬은 RNA 서열에서 가능한 모든 염기쌍에 대한 확률을 포함하고 있으며, 이는 많은 RNA 정보학 작업에 사용되어 왔습니다. 여기서는 염기쌍 결합 확률 외에도, 원래의 염기쌍 결합 확률 행렬 위에 역수, 역수의 제곱, 역수의 세제곱 쌍별 거리 행렬을 추가로 쌓습니다. 여기서 거리란 두 뉴클레오타이드 쌍 사이의 공유 결합(covalent bond)의 수를 의미하며(이는 RNA 그래프에서 공유 결합만 있는 경로 길이로도 간주될 수 있습니다). 역수 거리 행렬은 뉴클레오타이드 쌍 사이의 상대적인 거리에 대한 정보를 어느 정도 인코딩합니다. 뉴클레오타이드 쌍 사이에 공유 결합의 수가 적은 경우, 이들 사이의 거리가 공간적으로 더 가깝게 나타날 가능성이 큽니다. 거리 행렬이 이미 위치에 대한 정보를 인코딩하고 있기 때문에, 우리는 mRNA에 대해 위치 인코딩을 사용하지 않습니다.

RNAdegformer에서 사용되는 1차원 컨볼루션 연산은 패딩을 사용하지 않기 때문에, 컨볼루션 결과는 컨볼루션 커널 크기가 1보다 클 때 L 차원에서 차원이 축소됩니다. 그 결과, 염기쌍 결합 확률 행렬을 자기 주의 행렬에 직접 추가할 수 없습니다. 이를 해결하기 위해, 수정된 염기쌍 결합 확률 행렬에 대해 패딩 없이 동일한 커널 크기를 가진 2차원 컨볼루션을 수행하여, 특성 맵의 차원이 C×(L−k+1)×(L−k+1)이 되도록 합니다. 이제 주의 함수(attention function)는 다음과 같습니다:

개념적으로, 염기쌍 간의 상호작용 매핑 대신, 수정된 염기쌍 결합 확률 행렬의 2차원 컨볼루션 결과는 1차원 컨볼루션 K-머 결과와 일치하는 차원을 가진 K-머 간의 쌍별 상호작용 매핑으로 볼 수 있습니다. 차원 일치 외에도, 2차원 컨볼루션 연산은 mRNA 접힘의 기하학과 관련된 일부 누락된 정보를 보완해 줍니다. 이를 설명하기 위해, 우리는 OpenVaccine 데이터셋에서 mRNA 서열을 시각화하여 2차원 컨볼루션 연산의 물리적 및 수학적 이유를 설명합니다(Figure 2A).
A-20(위치 20의 A), G-21, C-40 및 U-41 간의 상호작용을 조사할 때, A-20과 C-40이 서로 상당히 가까이 있으며, 이들 간에 어느 정도의 상호작용이 있다고 상상할 수 있습니다. 비록 A-20과 C-40이 수소 결합을 형성하지 않더라도 말입니다. 그러나 A-20(위치 20의 A), G-21, C-40 및 U-41 간의 상호작용을 살펴보면, 염기쌍 결합 확률(BPP) 행렬과 거리 행렬의 해당 부분에서 이 정보가 전달되지 않는다는 것을 알 수 있습니다. 왜냐하면 (40, 20) 구성 요소는 BPP 행렬과 거리 행렬 모두에서 값이 0이거나 거의 0에 가깝기 때문입니다.
2×2 컨볼루션 커널이 BPP 행렬과 거리 행렬에서 작동할 때(여기서는 설명을 위해 모든 값을 동일하게 설정한 커널을 그립니다), 이는 본질적으로 A-20, G-21, C-40 및 U-41 간의 네 개의 연결을 융합하여 2×2 멀 (A-20, G-21 및 C-40, U-41) 간의 강한 연결을 생성합니다. 이제 네트워크가 A-20과 C-40 간의 상호작용(또한 G-21과 U-41 간의 상호작용)을 학습하는 것이 훨씬 쉬워집니다.

Figure 2: RNAdegformer는 컨볼루션과 자기 주의를 결합하여 RNA 분해를 예측합니다.
A. 생물물리 모델로부터 추가적인 입력 정보를 활용하는 RNAdegformer 아키텍처.
B. BPP+거리 행렬, 학습되지 않은 RNAdegformer의 주의 가중치, 그리고 학습된 RNAdegformer의 주의 가중치의 시각화.
컨볼루션과 자기 주의를 결합하는 것만으로는 뉴클레오타이드 위치별 예측을 생성할 수 없습니다. 이는 단일 염기쌍 인코딩 대신 K-머 인코딩을 생성하기 때문입니다. 뉴클레오타이드 위치별로 예측을 수행하기 위해, 우리는 추가적인 디컨볼루션(deconvolution) 레이어를 도입하여 전체 차원 인코딩을 복구합니다. 이를 통해 트랜스포머 인코더 전후에 1D 및 2D 인코딩의 잔여 연결(residual connections)을 허용합니다. 우리는 이러한 블록을 Conv-트랜스포머-인코더라고 명명합니다. 결과적으로, 단일 뉴클레오타이드 임베딩과 수정된 BPP 행렬 모두 예측을 출력하기 전에 심층 변환을 거칩니다.
이제 RNA 작업에 사용된 RNAdegformer 아키텍처를 요약할 수 있습니다(Figure 2B). 이는 여러 Conv-트랜스포머-인코더의 연속적인 특수한 경우로 볼 수 있습니다. 각각의 Conv-트랜스포머-인코더는 단일 트랜스포머 인코더 레이어 뒤에 디컨볼루션 레이어가 이어집니다. 또한 OpenVaccine 챌린지는 RNA 서열의 각 위치에서 예측을 수행해야 하므로, 마지막 트랜스포머 인코더 레이어는 K-머 인코딩 대신 단일 뉴클레오타이드 인코딩에서 작동하는 것이 중요합니다. 이러한 고려사항을 바탕으로, 우리는 ****와 ****라는 두 가지 주요 하이퍼파라미터를 동일하게 설정하여 RNAdegformer 스택을 구성하는 간단한 전략을 선택했습니다. 첫 번째 단일 레이어 Conv-트랜스포머-인코더는 **k**를 가지며, 다음 Conv-트랜스포머-인코더에서는 컨볼루션 커널의 크기를 1씩 줄입니다. 따라서 스택의 마지막 Conv-트랜스포머-인코더에 도달하면, ****는 1이 되며, 마지막 Conv-트랜스포머-인코더는 단순히 BPP 특징 맵에서 추가된 바이어스를 가지는 트랜스포머 인코더 레이어가 됩니다.
최적화기 및 학습 일정
처음에는 Adam 최적화기를 사용했지만, 이는 과소적합을 일으킨다는 것을 발견했습니다. 따라서 우리는 더 최근에 개발된 강력한 최적화기인 Ranger로 전환했습니다. 이 최적화기는 https://github.com/lessw2020/Ranger-Deep-Learning-Optimizer [49]에서 제공하는 그래디언트 중심화(gradient centralization)를 사용합니다. 학습 일정과 관련하여, 우리는 'flat and anneal' 방법을 사용했습니다. 학습은 1e-3의 평탄한 학습률로 시작하며, 전체 에포크 중 75%가 지나면 코사인 애닐링(cosine annealing) 스케줄을 사용하여 학습률을 0까지 줄이며 학습을 진행합니다. 가중치 감소(weight decay)는 0.1로 설정됩니다.
학습 접근 방식
OpenVaccine 챌린지에서는 테스트 세트의 샘플 수가 훈련 세트의 샘플 수를 초과하므로, 우리는 다양한 학습 방법을 결합하여 사용합니다(Figure 3). 여기서는 이러한 학습 방법에 대해 설명합니다.

Figure 3: RNAdegformer는 먼저 비지도 학습 환경에서 사전 학습된 후, 지도 학습 환경에서 실제 레이블(ground truth labels)로 훈련되며, 마지막으로 반지도 학습 환경에서 가짜 레이블(pseudo labels)과 실제 레이블을 함께 사용하여 훈련됩니다.
멀티태스킹 학습:
사전 학습(pretraining) 동안 변형되거나 마스킹된 서열, 구조 및 예측된 루프 유형이 RNAdegformer에 입력되며, RNAdegformer는 교차 엔트로피 손실(crossentropy loss)을 통해 RNA 서열의 각 위치에서 올바른 서열, 구조 및 예측된 루프 유형을 동시에 복구하도록 학습됩니다. 실제 레이블(ground truth labels)과 가짜 레이블(pseudo labels)로 훈련할 때는, RNAdegformer는 RNA 서열의 각 측정된 위치에서 다섯 가지 다른 분해 특성을 동시에 예측하도록 학습됩니다.
비지도 학습 (사전 학습):
우리는 OpenVaccine 챌린지 데이터셋에서 사용 가능한 모든 서열을 사용하여, 무작위로 변형되거나 마스킹된(빈 토큰과 함께) 서열 복구 손실(기본적으로 소프트맥스를 사용하여 올바른 뉴클레오타이드/구조/루프를 복구)을 목표로 네트워크를 사전 학습합니다. 사전 학습 동안 RNAdegformer는 생물물리 모델에 의해 제공되는 생물물리 지식을 바탕으로 RNA 구조의 규칙을 학습합니다.
지도 학습:
지도 학습 중에는 RNAdegformer가 RNA 분해 특성의 목표 값(target values)을 기준으로 훈련됩니다.
반지도 학습:
RNA에 대한 지도 학습 후, RNAdegformer는 서로 다른 깊이를 가진 RNAdegformer의 앙상블이 생성한 가짜 레이블을 사용하여 반지도 방식으로 재훈련됩니다. 반지도 학습에 대한 이전 연구와 유사하게【50】, 우리는 먼저 평탄한 학습률에서 가짜 레이블을 사용하여 모델을 재훈련하고, 그 후에 코사인 애닐 스케줄을 사용하여 훈련 세트에서 실제 레이블로 미세 조정합니다.
실제로, 우리는 먼저 비지도 학습으로 신경망을 사전 학습한 후, 지도 학습으로 실제 레이블을 사용하여 신경망을 훈련하며, 마지막으로 반지도 학습을 통해 레이블이 있는 데이터와 없는 데이터를 함께 사용합니다.

표 1:
OpenVaccine 데이터셋에서 mRNA 분해를 예측하기 위해 서로 다른 생물물리 모델로 생성된 입력을 사용한 성능.

표 2:
RNAdegformer 예측값과 시험관 내 반감기(in vitro half-life) 간의 피어슨 상관계수(Pearson correlation)를 다른 최고 방법들과 비교한 결과.
오류 가중 손실(Error weighted loss):
우리는 RMSE(평균 제곱근 오차) 손실의 한 형태를 사용했으며, 이는 수학적으로도 대회에서 사용된 지표와 동일합니다:

여기서 N_t는 열의 수, n은 예측된 위치의 수, y는 실제 값(ground truth), y^는 예측된 값(predicted value)을 의미합니다. OpenVaccine 데이터셋은 오류가 있는 실험적 측정값에서 나온 것이기 때문에, 우리는 지도 학습 중 각 측정값에 대한 오류를 기반으로 손실을 조정했습니다.

여기서 손실(loss)은 역전파(backpropagation)될 위치별 손실을 의미하며, 오류(error)는 해당 위치에 대한 실험적 오류를 나타냅니다. 그리고 α와 β는 실험적 오류에 따라 손실이 어떻게 가중되는지를 제어하는 조정 가능한 하이퍼파라미터입니다.
만약 α를 1로 설정하고 β를 무한대로 설정하면, 손실 값은 그대로 유지됩니다. 그렇지 않으면, 큰 오류를 가진 측정값에서의 그래디언트는 낮아져서 신경망이 실험적 오류에 과적합되는 것을 방지할 수 있습니다. 우리는 최적의 설정값이 α=0.5와 β=5임을 발견했습니다.
훈련 중 생물물리 모델의 사용
우리는 RNAsoft [11], rnastructure [12], CONTRAfold [9], EternaFold [51], NUPACK [10], 그리고 Vienna [8]를 포함한 생물물리 모델 앙상블에 의해 예측된 이차 구조를 사용했습니다. Arnie(https://github.com/DasLab/arnie)는 이차 구조 예측을 생성하기 위한 래퍼(wrapper)로 사용되었습니다. 각 서열에 대해, 우리는 37°C와 50°C에서 이차 구조 예측을 생성했는데, 이는 두 가지 평가된 분해 특성이 다른 온도에서 측정되었기 때문입니다. 비록 우리는 pH 10에서의 분해 특성에 대한 예측도 해야 했지만, 사용된 생물물리 모델 중 어느 것도 다른 pH에서의 예측을 생성할 수 없었습니다. 여섯 개의 패키지를 사용하여, 각 서열에 대해 12개의 이차 구조 예측을 얻었습니다. 훈련 중에는, 순방향 및 역방향 전파(pass) 과정에서 각 샘플에 대해 12개의 이차 구조 예측 중 하나를 무작위로 선택했습니다. 검증 및 테스트 중에는, 12개의 이차 구조 예측으로 생성된 평균 예측값을 사용했습니다.
최적의 하이퍼파라미터


Figure 4
RNAdegformer는 뉴클레오타이드 수준에서 RNA 분해를 정확하게 예측합니다.
A. 훈련 세트(파란색), 테스트 세트(주황색), 무작위로 생성된 세트(녹색)의 RNA 서열을 나타낸 T-SNE 플롯
B. BPP+거리 행렬, 학습되지 않은 RNAdegformer의 주의 가중치, 그리고 학습된 RNAdegformer의 주의 가중치의 시각화
C. OpenVaccine 대회 및 대회 후 실험에서 훈련된 사전 학습된 모델과 학습되지 않은 모델의 비교

Figure 5
OpenVaccine 비공개 테스트 세트에서 RNAdegformer의 R^2점수 (높아야 좋음)
A. 지도 학습만 수행된 경우,
B. 사전 학습된 경우,
C. 반지도 학습된 경우.
결과
자기 주의(Self-Attention)의 해석 가능성은 의사결정에 도움을 줍니다
대회가 끝날 무렵, 우리는 최종 제출에 사용할 두 가지 모델 세트를 훈련했습니다. 하나는 레이블이 있는 짧은 서열에 대해 직접 훈련된 모델이고, 다른 하나는 레이블이 있는 짧은 서열을 훈련하기 전에 사용 가능한 모든 서열(레이블이 없는 테스트 서열 포함)을 사용하여 사전 훈련된 모델입니다. T-SNE 분석에 따르면 테스트 서열은 다른 분포를 가지고 있는 것으로 보입니다(Figure 4A). 제출을 견고하게 선택하기 위해, 우리는 트랜스포머 인코더에서 학습된 주의 가중치를 시각화하고 평가했습니다(Figure 4B). BPP 행렬과 거리 행렬을 바이어스로 추가했기 때문에, 사전 훈련된 모델과 비사전 훈련된 모델 모두 학습된 주의 분포가 BPP와 거리 행렬을 닮았지만, 몇 가지 중요한 차이점도 있었습니다.
비사전 훈련된 모델은 주의 행렬의 대각선과 평행하게 나타나는 밝은 줄무늬로 표시된 것처럼, 위치적으로 가까운 뉴클레오타이드 쌍에 무분별하게 높은 주의를 기울였습니다. 이는 비사전 훈련된 모델이 mRNA 분해 특성 예측 시 위치적으로 가까운 뉴클레오타이드가 항상 중요하다고 생각했다는 것을 나타내며, 이는 매우 가능성이 낮아 보였습니다. 반면에, 사전 훈련된 모델은 위치적으로 가까운 뉴클레오타이드 쌍에 대한 같은 편향을 보이지 않았으며, 원래 BPP 행렬에서 거의 보이지 않는 약한 BPP 연결을 인식할 수 있었습니다. 이 경우, 모델은 생물물리 모델이 생성한 BPP 행렬을 더 효과적으로 활용했습니다.
이러한 고려사항과 대회의 짧은 시간 때문에, 우리는 사전 훈련된 모델을 실험하는 데 더 집중하기로 신속하게 결정해야 했으며, 최종 제출에서는 사전 훈련된 모델을 선호했습니다. 비공개 테스트 세트에서의 결과는 주의 가중치의 시각적 검토를 기반으로 한 우리의 선택을 확인해 주었습니다(Figure 4B). 사전 훈련된 모델은 공공 테스트 세트와 비공개 테스트 세트 모두에서 비사전 훈련된 모델보다 훨씬 더 좋은 성능을 발휘했습니다. 비사전 훈련된 모델을 사용했다면, 우리는 1636명 중 39위에 머물렀을 것이고, 실제로는 7위를 차지했습니다. 주목할 점은, 비공개 테스트 세트에서의 예측이 훨씬 더 큰 오류를 보였는데, 이는 아마도 비공개 테스트 세트에서 서열의 길이가 더 길고 서열의 다양성이 더 높기 때문일 것입니다.
반지도 학습이 RNA 분해 예측의 정확도를 높임
대회 이후, 우리는 비공개 테스트 세트에 대한 앙상블 예측에서 생성된 예측값을 반지도 학습 환경에서 가짜 레이블(pseudo-label)로 사용하면, 테스트 세트의 MCRMSE를 0.34198에서 0.33722로 줄일 수 있다는 것을 발견했습니다(Figure 4B). 이는 사용된 가짜 레이블의 예측값이 비공개 테스트 세트에서 0.3438의 점수만을 얻을 수 있었다는 점을 감안할 때 다소 놀라운 결과입니다.
지도 학습만 수행된 모델과 사전 학습된 모델, 반지도 학습된 RNAdegformer의 R^2 점수를 비교해보면, 사전 학습과 반지도 학습이 지도 학습만 수행된 경우에 비해 상당한 개선을 가져왔음을 알 수 있습니다(Figure 4C, Figure 5). 또한, deg_Mg_pH10에 대한 예측이 다른 두 가지 특성에 비해 상당히 큰 오류를 가지고 있다는 점을 주목할 필요가 있습니다. 이는 사용된 생물물리 모델들이 서로 다른 pH에서의 이차 구조 예측을 생성할 수 없기 때문에 예상된 결과입니다.
또한, 반지도 학습 접근 방식은 가짜 레이블링과 테스트 세트 분포를 사전에 알아야 한다는 점을 유의해야 합니다. 따라서, 가짜 레이블링은 대회 환경에서는 효과적일 수 있지만, 실제 응용에서 성능 향상이 그대로 나타나지 않을 수 있는 위험한 접근 방식입니다.
덧붙여, 우리는 다양한 패키지를 사용하고 앙상블한 모델의 성능도 보고합니다(표 1). 전반적으로 RNAsoft와 Vienna가 일관되게 최고의 성능을 제공하지만, 여러 생물물리 모델에서 앙상블한 결과가 개별 생물물리 모델보다 여전히 이점이 있습니다.

Figure 6
RNAdegformer는 더 긴 mRNA 서열에 대해 잘 일반화되며, mRNA 분해를 결정하는 중요한 특징을 드러냅니다.
A. RNAdegformer 예측값의 반감기(half-life) 상관관계를 이전의 최고 방법들과 비교한 결과.
B. OpenVaccine 데이터셋에서 LOFO(Leave-One-Feature-Out) 기능 중요도 분석.
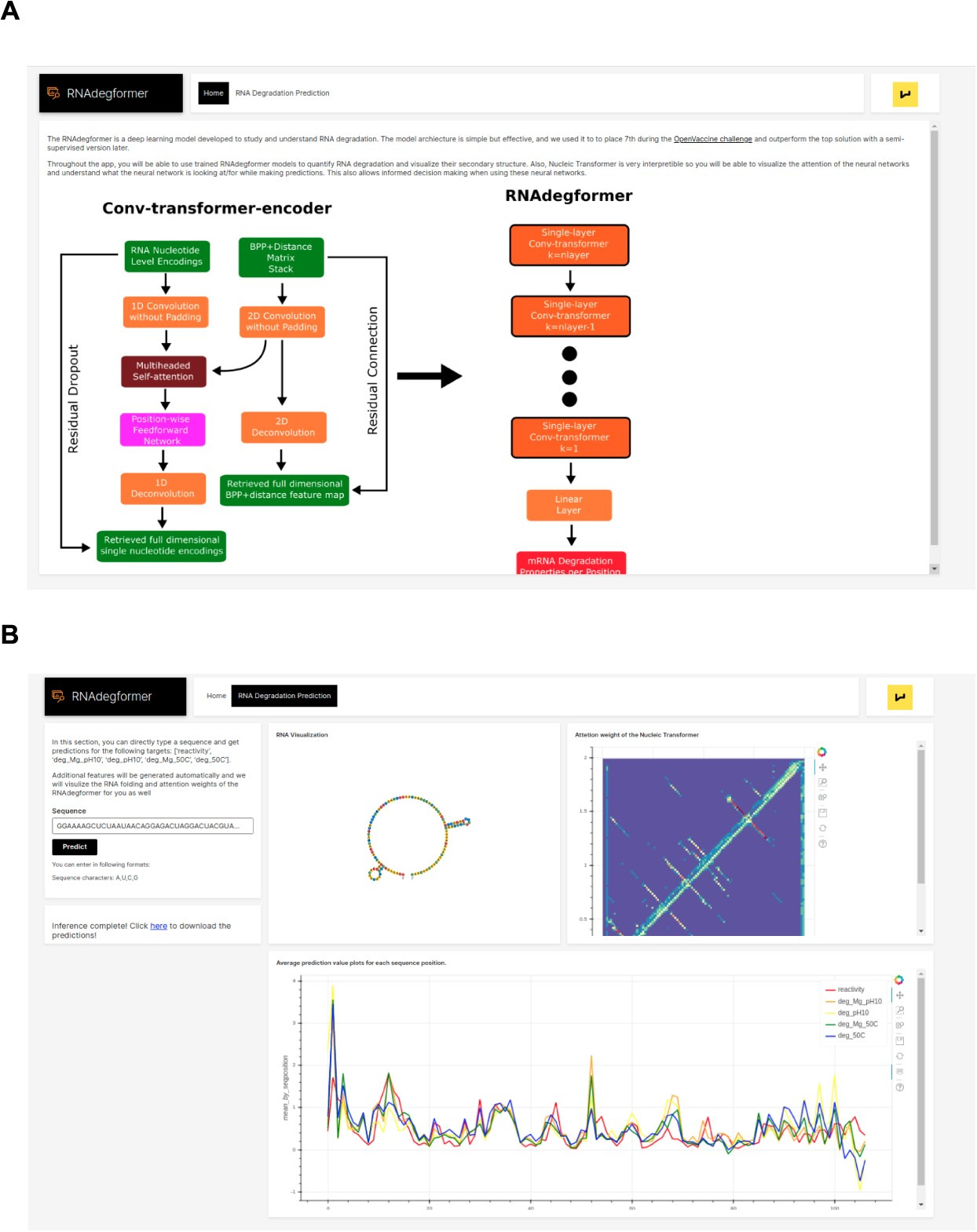
Figure 7
RNAdegformer 웹 애플리케이션
A. RNAdegformer 웹 애플리케이션의 홈 페이지.
B. 사용자가 이차 구조를 시각화하고, 어떤 RNA 서열에 대해서든 분해 속도를 예측할 수 있는 페이지.
RNAdegformer는 mRNA의 시험관 내 반감기를 예측하는 강력한 도구
RNAdegformer의 예측값을 CDS 변이체 그룹의 mRNA 설계 서열과 반감기 데이터【7】와 비교한 결과, RNAdegformer는 나노루시퍼라제 CDS 변이체의 가장 큰 그룹(평균 928 bp)의 반감기와 더 높은 상관관계(Pearson R=-0.655)를 보였습니다. 이는 mRNA 분해를 정량화하는 이전 모델들인 Degscore(Pearson R=-0.637) 및 비결합 확률 합(SUP, Pearson R=-0.58)과 비교하여 개선된 결과입니다(Figure 4D). 평균 1191 bp의 eGFP와 평균 505 bp의 다중 에피토프 백신(MEV) 같은 더 작은 CDS 변이체 그룹에서도 RNAdegformer는 Degscore(-0.288/-0.240)와 SUP(-0.103/-0.130)보다 큰 개선(-0.499/-0.578)을 보였습니다.
OpenVaccine에서의 최고 솔루션들과 비교했을 때, RNAdegformer는 훈련 데이터에 비해 훨씬 긴 CDS 변이체(Nanoluciferase 및 GFP)에 대해 더 잘 일반화된다는 것을 발견했습니다【2】. 전반적으로, RNAdegformer는 모든 3개의 CDS 그룹에서 -0.5 이상의 상관관계를 제공하는 유일한 방법입니다.
연구에 따르면, mRNA 서열의 CDS 영역을 컴퓨터 설계를 통해 최적화하면, Degscore【7】라는 능선 회귀 모델을 사용해 Gibbs 자유 에너지의 동적 프로그래밍 최적화를 통해 시험관 내 반감기를 최대 3배까지 증가시킬 수 있습니다. 우리는 RNAdegformer가 CDS 영역의 컴퓨터 설계를 안내하여 더욱 안정적인 서열을 생성할 수 있다고 믿습니다.
RNAdegformer는 RNA 안정성 예측에서 가장 중요한 특징을 밝혀냄
RNA 안정성을 예측하는 데 있어 가장 중요한 특징이 무엇인지 이해하기 위해, 우리는 간단한 LOFO(Leave-One-Feature-Out) 접근 방식을 사용했습니다. 우리는 최적의 설정으로 모델을 처음부터 재훈련했지만, 사용 가능한 특징 중 하나(서열, 예측된 구조, 예측된 루프 유형 또는 예측된 염기쌍 결합 확률)를 제외한 상태에서 모델 성능을 평가했습니다(Figure 4G).
그 결과, 서열 정보를 제외했을 때 모델 성능에 가장 큰 영향을 미쳤음을 발견했습니다. 이는 이차 구조 외에도 실험 데이터를 통해 안정적인 원시 서열 모티프/모티프 쌍을 추출하여 더 안정적인 RNA 분자를 설계할 수 있다는 것을 나타냅니다【7】. 또한, 이는 RNAdegformer가 모티프/쌍별 모티프 인식 측면에서 강력한 능력을 가지고 있음을 보여줍니다. 서열 정보를 제외했을 때의 성능 저하 다음으로는 특정 RNA 서열이 채택할 수 있는 가능한 접힘의 앙상블을 설명하는 예측된 염기쌍 결합 확률이 큰 영향을 미쳤습니다. 이는 염기쌍 결합 확률이 RNA 이차 구조를 잘 설명하는 풍부하고, 자세하며, 포괄적인 특징임을 보여줍니다.
또한, 실험적으로 염기쌍 결합 확률이 RNA 반감기와 높은 상관관계가 있다는 것이 입증되었으며【7】, 결합을 최대화하도록 RNA 코딩 서열을 설계하는 것이 더 안정적인 RNA 분자를 생성하는 전략으로 제안되었습니다【53】. 우리의 LOFO 결과는 이러한 제안을 뒷받침합니다. 더 나아가, RNAdegformer가 염기쌍 결합 확률과 같은 2D 특징을 직접적이고 효과적으로 통합할 수 있는 능력도 그 유연성을 보여줍니다. 놀랍게도, 구조와 루프 특징을 제외했을 때 모델 성능에 거의 영향을 미치지 않았으며, MCRMSE는 거의 동일하게 유지되었습니다. 그러나 구조와 루프 특징의 최소한의 영향은 염기쌍 결합 확률의 중요성에 관한 이전의 발견과 일치합니다. 구조와 루프 특징은 예측된 염기쌍 결합 확률에 기초하여 단일 가장 가능성 높은 접힘만을 나타냅니다. 즉, 염기쌍 결합 확률처럼 RNA 이차 구조를 포괄적으로 설명하지 않습니다. Degscore와 선형 설계는 구조 정보나 열역학을 활용하는 반면, 우리의 RNAdegformer는 mRNA 분해와 관련하여 가장 중요한 특징인 서열과 BPP를 동시에 효과적으로 활용할 수 있습니다. 이는 LOFO 소거 연구에서 밝혀진 바입니다.
RNAdegformer 웹 애플리케이션
우리는 H2O.ai의 Wave를 사용하여 개발된 웹 애플리케이션(Figure 7A)을 만들었으며, 이를 https://github.com/Shujun-He/RNAdegformer-Webapp 에서 사용할 수 있도록 했습니다. 이를 통해 사용자는 코딩 없이도 RNAdegformer를 사용하여 RNA 분해를 예측하고 시각화할 수 있습니다. 웹 애플리케이션을 사용하려면, 사용자가 관심 있는 RNA 서열을 입력하기만 하면 됩니다(Figure 7B). 그러면 RNA 분해 예측 결과가 다운로드 가능한 CSV 파일로 생성되며, BPP와 이차 구조와 같은 관련 특징도 함께 제공됩니다. 또한, RNA 서열의 이차 구조, 주의 가중치 및 분해 특성도 시각화되어 사용자가 분해 예측을 시각적으로 검사할 수 있습니다.
토론
이 연구에서 우리는 RNAdegformer라는 효과적인 신경망 아키텍처를 제시하며, 이는 mRNA COVID-19 백신 후보의 뉴클레오타이드별 분해 특성을 정확하게 예측할 수 있습니다. 우리는 최근 21일간의 OpenVaccine 챌린지에 RNAdegformer를 적용하여 참여했으며, 전 세계의 최고 수준의 머신 러닝 전문가 1636팀 중에서 7위를 차지했습니다. 또한, 반지도 학습을 통해 RNAdegformer가 OpenVaccine 챌린지에서 1위를 차지한 솔루션을 상당한 차이로 능가함을 보여주었습니다. RNAdegformer는 이전의 최고 방법들보다 더 나은 상관관계를 보이며, 보지 못한 mRNA 서열의 반감기를 예측하는 데에도 잘 일반화됩니다. 더 나아가, RNAdegformer는 RNA 분해를 예측하는 데 가장 중요한 특징들을 밝혀냅니다. 우리의 결과는 자기 주의(self-attention)와 컨볼루션(convolution)이 전역적(global) 및 지역적(local) 의존성을 모두 효과적으로 학습하여 RNA 분해와 반감기를 예측하는 데 강력한 조합임을 보여줍니다. 트랜스포머 아키텍처가 자연어 처리(NLP) 이상으로 뛰어날 수 있다는 것은 오래전부터 제기되어 왔으며, 우리의 연구는 이를 입증합니다.
비록 mRNA 이차 구조를 예측하는 많은 동적 프로그래밍 알고리즘이 존재하지만, 뉴클레오타이드별로 mRNA 분해 특성을 예측하는 것은 전례가 없었습니다. COVID-19로 인해 혼란스러웠던 2020년 이후, mRNA 백신은 COVID 문제에 대한 빠르고 효과적인 해결책으로 등장했으며, Pfizer와 Moderna 같은 회사들이 전례 없는 속도로 mRNA 백신을 출시했습니다. 그러나 취약한 mRNA 백신의 보관 및 운송은 여전히 과제입니다(Pfizer-BioNtech의 백신은 −70°C-, Moderna의 백신은 −20°C에서 보관해야 합니다). mRNA 가수분해를 줄이는 한 가지 전략은 동일한 단백질을 코딩하면서도 이중 가닥 영역을 형성하도록 RNA를 재설계하는 것입니다. 이중 가닥 영역은 이러한 분해 과정을 방지합니다【7, 53】. 우리의 연구는 더 안정적인 mRNA 백신 개발에 있어 가이드를 제공하고, 훈련된 모델이 스크리닝 도구로 작용할 수 있을 것입니다. 메신저 RNA 백신과 치료제는 감염병과 암에 많은 응용이 가능하며【54–57】, 우리는 RNAdegformer가 현재 백신보다 더 가혹한 조건을 견딜 수 있는 더 안정적인 mRNA 백신 설계에 도움이 될 것이라고 기대합니다.
그러나 중요한 점은, OpenVaccine 공공 세트의 107 bp mRNA 서열과 비공개 세트의 130 bp mRNA 서열 간에 상당한 오류 차이가 있으며, 이는 서열 길이와 다양성의 차이 때문입니다. 실제 COVID-19 후보는 더 길며, 이를 모델링하는 것은 미래의 과제로 남아 있습니다. 이러한 긴 서열의 경우, 자기 주의의 이차적 계산 복잡성(quadratic computational complexity)이 긴 서열에 대한 훈련을 금지하는 주요 도전 과제입니다. 주목할 만하게도, 최근에는 자기 주의의 이차적 계산 복잡성을 선형으로 줄여 훨씬 더 긴 서열에 대한 트랜스포머와 같은 자기 주의 훈련을 가능하게 하는 연구가 많이 진행되었습니다【58–61】. 전체 유전체와 COVID-19 mRNA 백신 모두 전체 자기 주의의 길이 제한을 크게 초과하며, 특히 COVID-19 백신 후보는 약 4000 bp 길이입니다. 따라서 이러한 새로운 접근 방식은 이러한 과제를 해결하는 효과적인 도구가 될 수 있습니다.
요약하자면, 우리는 mRNA 분해와 반감기 예측을 위한 컨볼루션과 트랜스포머 기반의 딥러닝 플랫폼을 개발했습니다. 우리의 연구는 RNA 안정성과 반감기 예측에서 성공을 거두었으며, 앞으로 추가적인 개발과 최적화를 통해 RNA 분해와 구조 관계를 이해하고, 차세대 mRNA 치료제 개발을 돕는 등 많은 도전을 해결할 수 있을 것이라고 믿습니다.
핵심 요점
- 메신저 RNA 치료제는 모듈성을 제공하고, 이론적으로는 어떤 단백질이든 전달하고 번역할 수 있는 매우 유망한 플랫폼으로 떠오르고 있습니다. 메신저 RNA는 시험관 내 전사(in vitro transcription)를 통해 빠르고 유연하게 생산될 수 있지만, 인라인 가수분해로 인해 화학적 불안정성 문제가 있습니다.
- 우리는 컨볼루션과 자기 주의(self-attention)를 활용하여 지역적(local) 및 전역적(global) 의존성을 모두 포착할 수 있는 모델 아키텍처인 RNAdegformer를 제시합니다. 이를 통해 mRNA 서열의 분해 특성을 예측하는 데 높은 정확도와 해석 가능성을 제공합니다.
- 비지도 학습(사전 학습), 지도 학습, 반지도 학습을 서로 결합하여 사용함으로써, RNAdegformer가 주어진 RNA 서열의 각 위치에서 RNA 분해율을 예측하는 데 있어 OpenVaccine의 최고 솔루션을 능가함을 입증합니다. 이는 안정적인 mRNA 백신과 치료제를 예측하고 생산하는 데 매우 중요한 작업입니다.
- RNAdegformer는 훈련 데이터셋보다 훨씬 긴 서열의 반감기를 예측하는 데 있어서 다른 머신러닝 및 동적 프로그래밍 알고리즘보다 더 잘 일반화됩니다.
- RNAdegformer는 leave-one-feature-out(LOFO) 테스트를 통해 mRNA 분해 예측에서 중요한 특징을 밝혀내어 RNA 분해에 대한 이해를 진전시킵니다.
저자 기여 성명
- S.H.와 B.G.는 프로젝트를 구상했습니다. R.S.는 RNA 서열의 분석과 생물학적 맥락에 대한 중요한 피드백을 제공했습니다. S.H.는 딥러닝 알고리즘을 구현했고(OpenVaccine 챌린지에 참여), Q.S.는 프로젝트를 감독하고 지침을 제공했습니다. S.H., B.G., R.S.는 Q.S.와의 협의를 통해 원고를 작성했습니다.
데이터 가용성
- OpenVaccine 데이터셋은 Kaggle에서 사용할 수 있습니다. 사전 학습된 모델은 Kaggle 노트북에서 액세스할 수 있습니다.
코드 가용성
감사의 말
- 우리는 RNA 분해에 대한 데이터를 제공하고 OpenVaccine 대회를 주최해 준 Das Lab에 감사드립니다. 또한 Dr. Yang Shen, Dr. Shuiwang Ji 및 그의 학생 Hao Yuan에게 원고를 교정해 주신 것에 대해 감사드립니다.
자금 지원
- National Institutes of Health (R01AI165433); Texas A&M University X-grants.
저자 소개
- Shujun He는 텍사스 A&M 대학교의 Artie McFerrin 화학공학과 박사과정 학생입니다. 그의 연구 관심사는 딥러닝과 mRNA 안정성입니다.
- Baizhen Gao는 텍사스 A&M 대학교의 Artie McFerrin 화학공학과 박사과정 학생입니다. 그의 연구 관심사는 유전자 회로 설계와 단백질 공학입니다.
- Rushant Sabnis는 텍사스 A&M 대학교의 Artie McFerrin 화학공학과 박사과정 학생입니다. 그의 연구 관심사는 단백질 공학과 합성 생물학입니다.
- Qing Sun은 텍사스 A&M 대학교의 Artie McFerrin 화학공학과 조교수입니다. 그녀의 연구 관심사는 생의학 및 환경 응용을 위한 합성 생물학입니다.
-----------------------------------------------------------------------------------------------------------------------------------------------------------------
참가했을때, 이 논문을 알았다면 더 좋지 않았을까...
https://www.kaggle.com/competitions/stanford-ribonanza-rna-folding/discussion/460203
Stanford Ribonanza RNA Folding | Kaggle
www.kaggle.com
https://github.com/tattaka/stanford-ribonanza-rna-folding-public
GitHub - tattaka/stanford-ribonanza-rna-folding-public: Stanford Ribonanza RNA Folding 4th place solution
Stanford Ribonanza RNA Folding 4th place solution. Contribute to tattaka/stanford-ribonanza-rna-folding-public development by creating an account on GitHub.
github.com
https://github.com/Shujun-He/RNAdegformer
GitHub - Shujun-He/RNAdegformer
Contribute to Shujun-He/RNAdegformer development by creating an account on GitHub.
github.com



